Molecular surveillance of SARS-CoV-2
Distribution of SARS-CoV-2 by major lineage

The table displays the distribution of SARS-CoV-2 lineages detected during weeks 2023/19 to 2023/26 in Luxembourg. This enables to approximately estimates the circulation of SARS-CoV-2 variants in Luxembourg.
SARS-CoV-2 Variant distribution by week in 2023
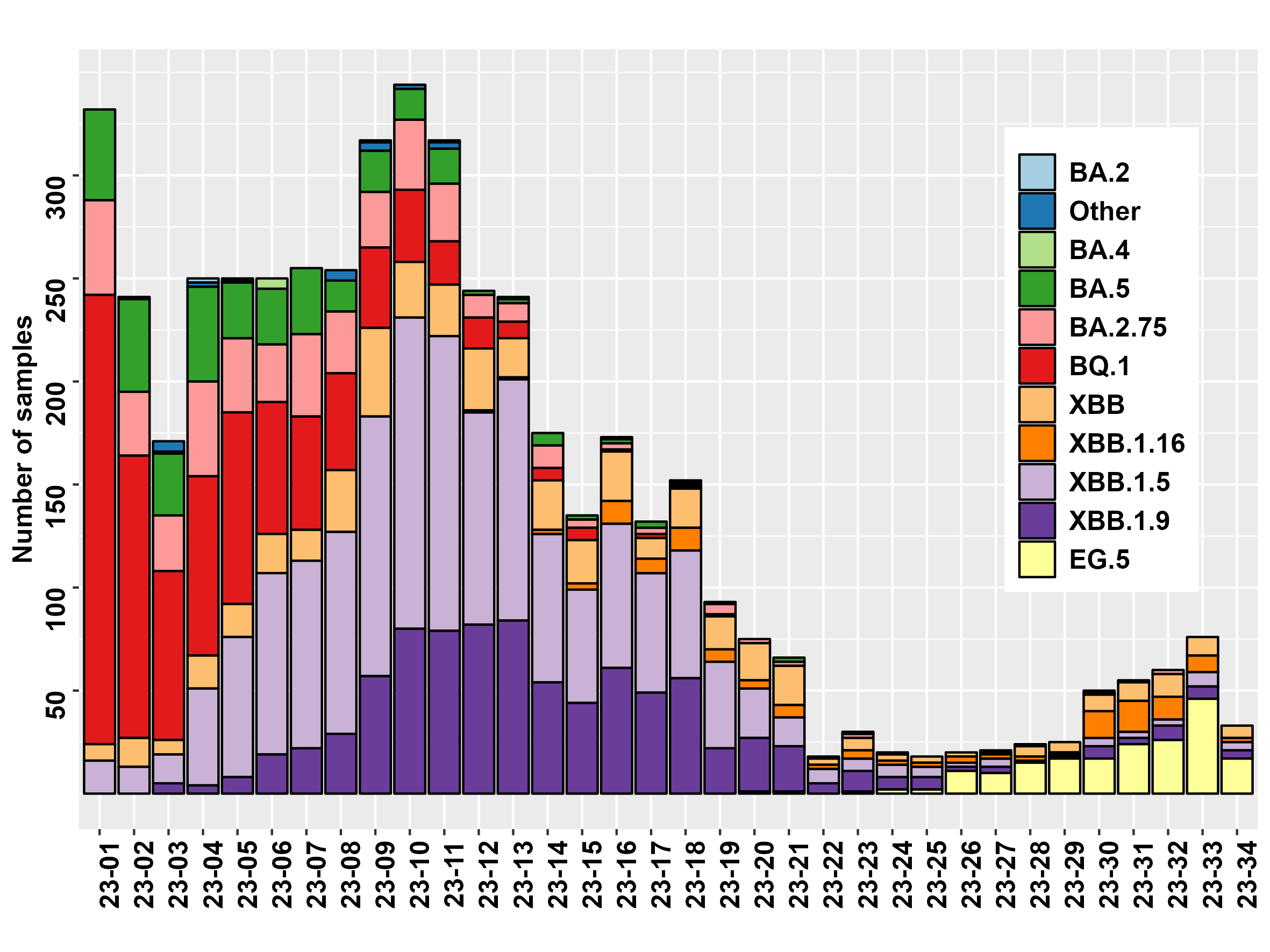
This figure shows the distribution of each major lineage by calendar week since beginning 2023. Since the end of February, XBB (including XBB.1.5 and XBB.1.9) variants have been responsible for the highest number of cases.
Population pyramid by major lineage
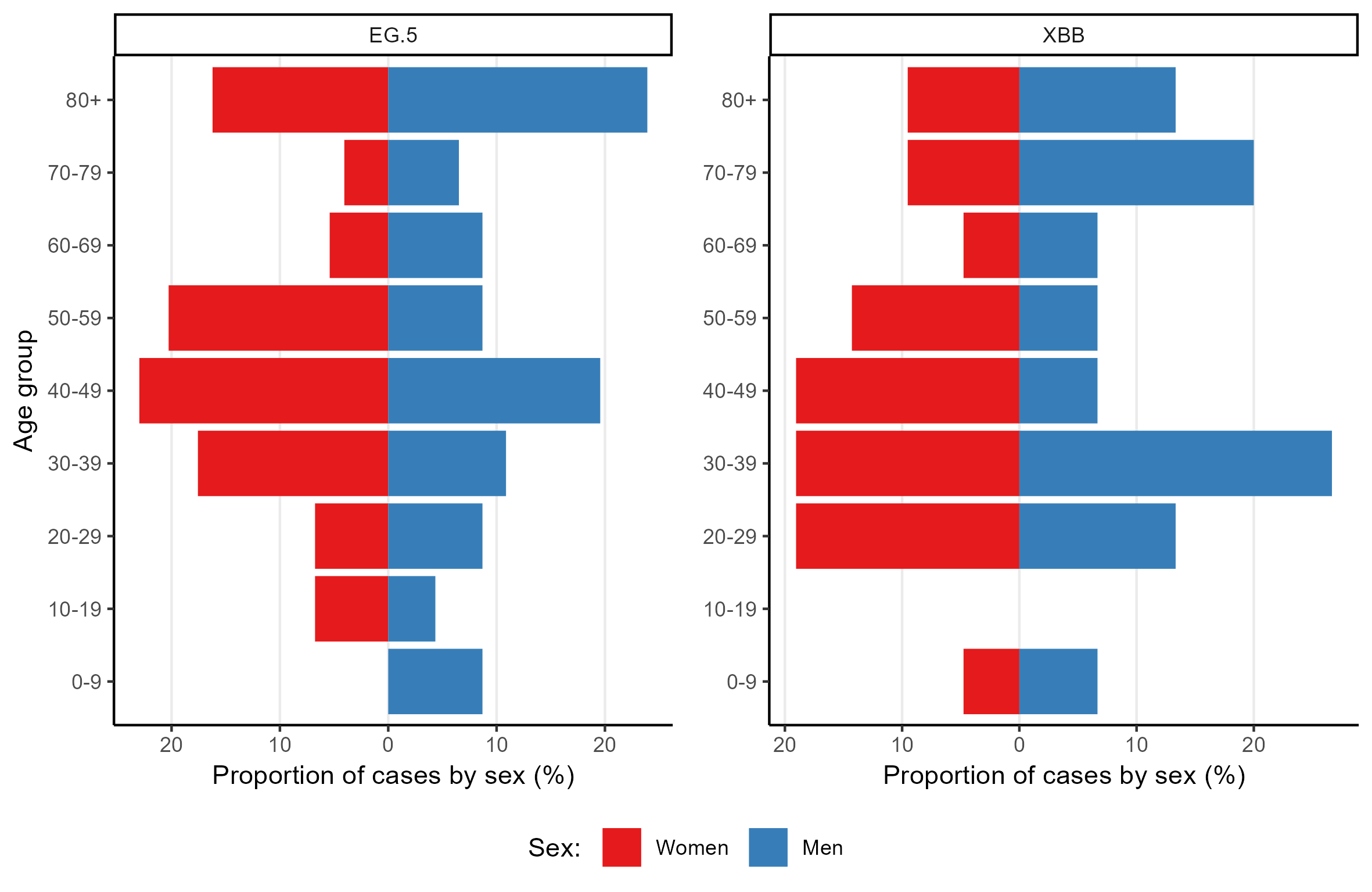
This figure displays the age group and sex distribution of specimens sequenced over the last 4 weeks, by lineage.
About COVID-19 Genomics in Luxembourg
Genomics is the study of the genome, i.e. the genetic sequence information of an organism and attempts to characterize and understand the structure and function of this information. Sequencing is the main technique for reading and interpreting this genetic information. It is used to determine the order of nucleotides in a complete genome.
Virus are known to undergo mutations, corresponding to errors in the genetic code, during their replication process. While some are harmless, others can bring about such changes in the virus that can make it more transmissible or deadly. Therefore, itis important to keep track of the genomic sequences in order to verify the virulence of the virus.
The genetic information of SARS-CoV2 corresponds to a genome of about 30,000 nucleotides. In order to be able to sequence quickly the complete genome of this virus to collect all relevant information, Next Generation Sequencing (NGS) providing large amounts of sequence data is the best adapted technique. Thus, the result of the whole genome sequencing (WGS) and the identification of specific mutations that characterised a variant allows to classify a virus as a particular variant and to determine its lineage affiliation. Moreover, NGS is a valuable technology not only because it can track the prevalence of known mutant strains but also because it can identify novel mutations, so novel variant.
Genomic surveillance consists to analyze similarities and differences among the whole genome sequences obtained for each specimen in a population over a defined timeframe. It allows visibility on the variants that circulate and if necessary to adapt the health measures taken according to these epidemiological data. This can help indicate routes of transmission enable identification and investigation of clusters and help inform strategies to control the spread of the virus. This real-time surveillance is possible by rapidly sequencing and sharing SARS-CoV2 genomic data.
Our LUX-GEMM unit plays a key role in the fight against Covid19 and this since the beginning of the epidemic. From early 2020, the laboratory rapidly implemented an in-house next generation sequencing protocol with Illumina technology adapted for whole-genome sequencing of SARS-CoV2. This could be done quickly thanks to our long-term experience in microbial sequencing and with an adapted technical platform and bioinformatics expertise. In April 2020, the first sequencing results were already delivered.
Thanks to this expertise and the implementation of our sequencing process and strategy, we are able to perform whole genome sequencing of the virus on a large selection of positive samples and to report these results regularly allowing visibility of SARS-CoV-2 mutations and variants through time and space in Luxembourg.
The Illumina technology corresponding to the sequencing by synthesis of small fragments of DNA is the first technique that has been implemented in our lab from the beginning of the crisis. The extracted RNA is converted to complimentary DNA. After amplification, purified and quantified DNA is cut up into fragments, end-repaired and A-tailed. Then, short segments of genetic material called adapters are ligated to the DNA fragments. This step is called “Tagmentation”. After clean-up, sequences from each sample are identified with a specific couple of indexes and after clean-up of the final libraries, the samples are pooled then quantitated and normalized before loading onto a flow cell adapted to the sequencer used. It is possible with this technology to pool several samples at the same time (for example max 384 on Next-Seq-1000). The preparation of the DNA and the library for sequencing take around two working days. The preparation of the library is a step that can be realized manually or by automation using the Hamilton NGS star device. Once the sequencer is started, the sequencing reaction on its own takes 20 to 24 hours.
In 2022, in parallel to Illumina technology, we have implemented a new technology using a third generation sequencer, the Gridion from Nanopore (ONT for Oxford Nanopore Technologies). The sequencing is done by passing the entire DNA molecule through an electrical pore in the sequencer. The preparation for sequencing is performed using the Rapid/Midnight SARS-CoV2 kit from Nanopore. After conversion of the RNA into DNA and amplification of different overlapping segments of the genome, each amplicon is barcoded with a unique barcode per sample. This allows the pool of different samples on one run. After clean-up and quantification, each fragment is linked to an adapter allowing the passage through the pore. Sample preparation for sequencing can be done within one day and the sequencing reaction on Gridion takes 20 hours.
After launching each sequencer, a large amount of data (up to millions of sequences of letters/nucleotides) which are then assembled together and aligned with a reference sequence. Then, bioinformatics programs compare the new sequence data to the reference sequence and identify variations in the genome sequenced. To assign a lineage to a genome according to these variants, our bioinformatic team use their pipeline updated regularly with the last version of Pangolin available. Then, these data are shared on public scientific database such as GISAID, so internationally available.
We have gone from a capacity of 380 at the end of 2020 to 1200 at the end of 2021 and in the course of 2022, our maximum capacity can reach at least 2500 sequencing per week. This can be achieved by the automatisation part of the Illumina sequencing samples preparation and also the implementation of the new Nanopore technology in parallel to the already installed Illumina technology.
We are also evolving by working on new sequencing approaches by the implementation of metagenomics and target enrichment approaches. These broadly approaches will enable us to have more information and to be more specific in the diagnosis of certain infections, for example in the case of emergence of a new virus or in the diagnosis of co-infections.